spora
Outputs
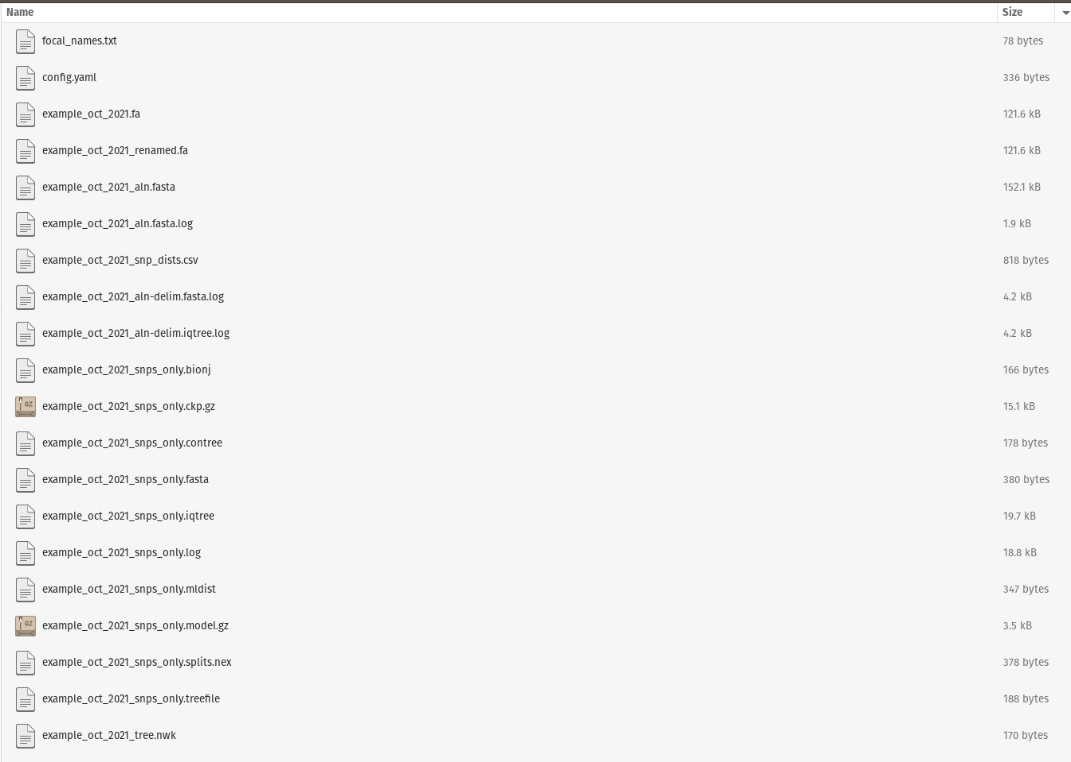
Currently spora creates its various output files all within the specified target output directory. The output files would appear as follows:
• {prefix}.fa - multi-FASTA (not aligned) that contains all focal sequences, as well as background sequences, if they are included.
• {prefix}_aln.fasta - multi-FASTA alignment (using MAFFT) of the above FASTA file.
• {prefix}_snp_dists.csv - SNP distance matrix of all samples included in the multi-FASTA. Generated from snp-dists using the multi alignment file with molten format selected and CSV format output. Note that only ACGT differences are counted (N’s are disregarded).
• {prefix}.nwk - phylogenetic tree in Newick format generated using augur tree (a wrapper for iqtree).
• {prefix}_snps_only.fasta - multi-FASTA file for all samples listed above, reduced just to positions that are variable in at least one of the samples, as compared to the rest of the FASTA contents. Generated using snp-sites and considers only SNPs with ATCG (N’s are disregarded).
• {prefix}_snps_only.contree - Consensus tree generated using the snps only FASTA described above. Created using iqtree2 with 1000 bootstraps.
• {prefix}_snipit.jpg - A jpg image that summarizes the SNP diversity among input sequences relative to the reference. generated using the software found here.
• {prefix}_summary_report.html - If --report is selected, then an HTMl summary report will be created in the output directory. This will contain basic information on run parameters and inputs, SNP diversity, and basic phylogenetics. Please see below for more information on the summary report.
For the outbreak described above with the prefix example_oct_2021, the output directory should have a similar output as shown below. Note that this directory structure also holds the config.yaml and focal sequence list in the output directory:
Summary output report
As of spora v0.5.0, there is the option to create a summary report in HTML format. The report contains the following sections:
• Summary Statistics
◦ Input sequences
◦ Retained sequences for analysis
• SNP diversity relative to the sequence
• SNP Distances
◦ SNP summaries relative to focal sequences
◦ SNP heatmap (all sequences retained for analysis)
• Phylogenetic trees